以p53-MDM2为靶点开发全新机制的抗肿瘤药物,是当前抗肿瘤药物研发领域的重点。目前基于p53-MDM2靶点尚未有上市药物,其面临的主要问题是:MDM2被抑制后,细胞受负反馈调节会使MDM2蛋白上调,因此需要更高浓度的抑制剂来达到原先抑制水平,而高浓度抑制剂会引起毒副作用,包括嗜中性粒细胞减少症、热性嗜中性粒细胞减少症、长期细胞减少症、骨髓衰竭和胃肠毒性等。因此,需要研发更有效的p53-MDM2阻断分子来克服上述问题。
蛋白降解靶向嵌合体(Proteolysis targeting chimera, PROTAC)是当前新兴的抗肿瘤药物研发技术,具有低剂量高效降解靶蛋白的优势。然而,PROTAC除了结合目标蛋白之外通常会引入第二靶点—E3泛素连接酶,当结合E3泛素连接酶时可能对第二靶点蛋白产生抑制作用或脱靶效应,从而引起相关毒副作用。有研究显示,利用含CRBN的E3配体设计的PROTAC除了降解目标蛋白外,还可同时降解其它蛋白导致脱靶效应等。同型PROTAC是PROTAC技术的一种新形式,由两个相同配体分子直接连接而成,即E3连接酶配体(靶蛋白配体)以二聚化形式存在以介导其自身结合蛋白的降解。与传统PROTAC分子相比,同型PROTAC的优势在于不引入第二靶点,可有效避免潜在的毒副作用。MDM2蛋白既是靶蛋白也是E3泛素连接酶,为同型PROTAC的设计提供了理论依据。
在前期设计的新型p53-MDM2/HDAC双靶点抑制剂(J. Med. Chem., 2018, 61, 16,7245-7260)研究工作基础上,我院何世鹏副研究员联合美国University of Arizona王卫教授成功设计了第一代MDM2蛋白Homo-PROTAC降解分子。当同型PROTAC结合MDM2蛋白时,一端配体连接的MDM2发挥E3泛素连接酶作用,给另一端的MDM2蛋白贴上泛素化“标签”,诱导MDM2蛋白进行“自杀式”降解,并上调p53蛋白。
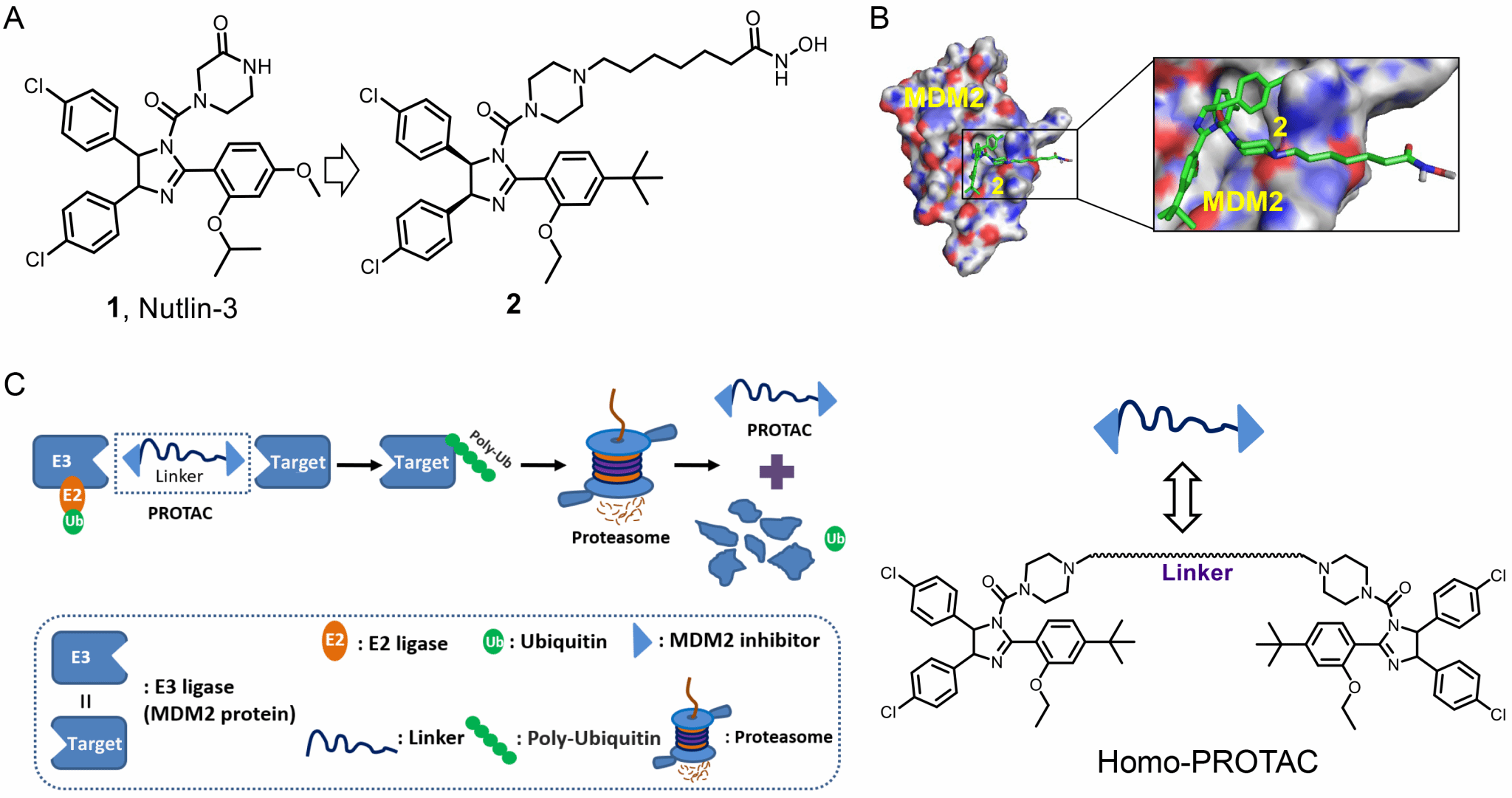
图1. Homo-PROTAC设计思路
生物活性结果显示,化合物11a对MDM2(Ki = 96 nM)竞争性抑制活性最强,并且对人肺癌A549细胞表现出较强的抑制活性。通过Western Blot实验在细胞水平上验证了该化合物通过蛋白酶体途径降解MDM2蛋白,上调p53蛋白,并引起A549肿瘤细胞凋亡。将11a进行手性拆分,得到的光学纯化合物11a-1显示出更为高效的MDM2蛋白降解活性。体内裸鼠A549移植瘤实验结果表明,化合物11a-1在腹腔注射给药剂量20 mg/kg下抑瘤率为45.6%,该化合物具有明确的量效关系,当腹腔注射给药剂量提高至30 mg/kg时,其抑瘤率达到52.4%,实验动物体重无明显变化。药代动力学实验表明,11a-1具有较为合理的半衰期和血药浓度时下分布曲线值(AUC),是一种具有具有良好PK/PD性能的新型PROTAC分子,目前进一步的结构优化研究正在进行中。